
第一作者和单位:杨志文 南开大学化学学院 元素有机化学国家重点实验室
通讯作者和单位:何良年 南开大学化学学院 元素有机化学国家重点实验室
原文链接:https://pubs.rsc.org/en/Content/ArticleLanding/2022/CY/D2CY01195F
关键词:电催化耦合,二氧化碳还原,5-羟甲基糠醛氧化,非共价负载分子催化,生物质转化
全文速览
南开大学何良年绿色化学团队建立了一种可由清洁太阳光驱动的电催化二氧化碳还原反应(ECO2RR)与5-羟甲基糠醛氧化反应(HMFOR)耦合电解体系。本工作中,合成了具有大空间位阻的酞菁钴分子TBP-CoPc和芘修饰的TEMPO分子(Py-TEMPO),将其非共价负载于碳纳米管(CNTs)上后,能分别在合适电位下以大于95%的法拉第效率(FE)电催化CO2还原至CO和5-羟甲基糠醛(HMF)氧化至2,5-呋喃二甲酸(FDCA)。通过将此氧化及还原反应于一个单独的电解池中组装,探究了耦合反应在分别控制阴极和阳极电位的三电极体系以及控制槽压的两电极体系中的表现。结果表明,阴、阳两极电位变化对于其产物分布具有重要影响,并且通过改变电极面积以及槽压的方式可以获得理想的电位区间。该耦合体系在2.5 V的槽压下,能以96.9% FE得到CO,以90.8% FE得到FDCA。该耦合电解系统可利用光伏发电驱动,阴极能够以超过50000的TON值及95.4%的选择性获得CO产物,同时阳极FDCA的产率可以达到69.9%。

背景介绍
大气中CO2浓度的不断增加和化石资源的枯竭已经引起了人们对未来全球环境的关注。电催化CO2还原可以通过利用清洁的电力资源,将主要的温室气体CO2还原为CO,HCOOH等储能小分子,有利于缓解温室效应以及化石能源短缺问题。常规CO2还原耦合的水氧化析氧反应,由于其动力学惰性及热力学稳定性,需要较多的能量投入且所得氧气的经济价值不高。同时,生物质利用也是节能减排的重要途径之一。生物质衍生的HMF(5-羟甲基呋喃甲醛)相对于水在热力学上更易发生氧化,且其氧化产物FDCA(呋喃二甲酸)可以替代来源于化石资源的对苯二甲酸生成多种聚酯材料,具有广阔应用前景。因此电催化CO2还原反应与HMF氧化反应的耦合电解不仅能够双向实现碳减排,而且可以降低能耗并提升整体电解产物的经济价值。

Fig. 1 Schematic illustration of the ECO2RR coupled with the HMFOR
研究目标
本文通过非共价负载方式构筑能够有效催化CO2还原及HMF氧化的分子基电催化剂,将其应用于耦合成对电解中并探究影响耦合效率的因素,旨在以清洁电力驱动耦合体系的高效运转。
图文精读
通过分子结构修饰,我们合成了具有大空间位阻的酞菁钴分子TBP-CoPc和芘修饰的TEMPO分子(Py-TEMPO),将其非共价负载于碳纳米管上后,得到了TBP-CoPc/CNTs及Py-TEMPO/CNTs催化剂,通过伏安线性扫描及循环伏安测试证实了其对于相应底物的催化转化活性。在恒电位电解实验中,TBP-CoPc/CNTs能够在−1.5 V(电位值均以饱和Ag/AgCl为参比)的电位下以96.2%的FE值催化CO2还原得到CO产物,而Py-TEMPO/CNTs也能在0.7 V ~ 0.9 V的电位区间内以> 95%的FE值高选择性催化FDCA产物的生成。
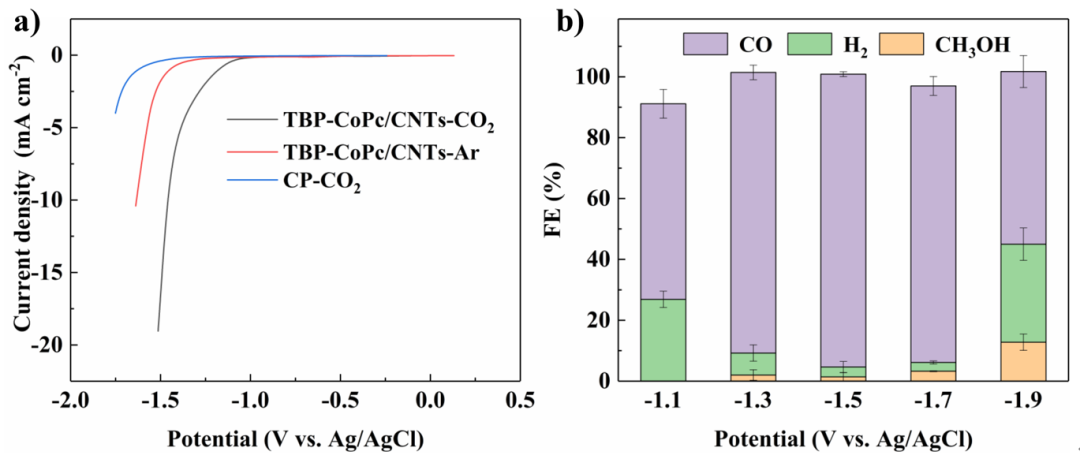
Fig. 2 Electrocatalytic activity of TBP-CoPc/CNTs for the ECO2RR. a) LSV curves of TBP-CoPc/CNTs under CO2 or Ar as well as the blank test with a bare carbon paper (CP) support under CO2; scan rate: 100 mV s−1. b) Faradaic efficiency for each reductive product under different potentials after 1 h of constant potential electrolysis. The average values and error bars are based on three parallel experiments.
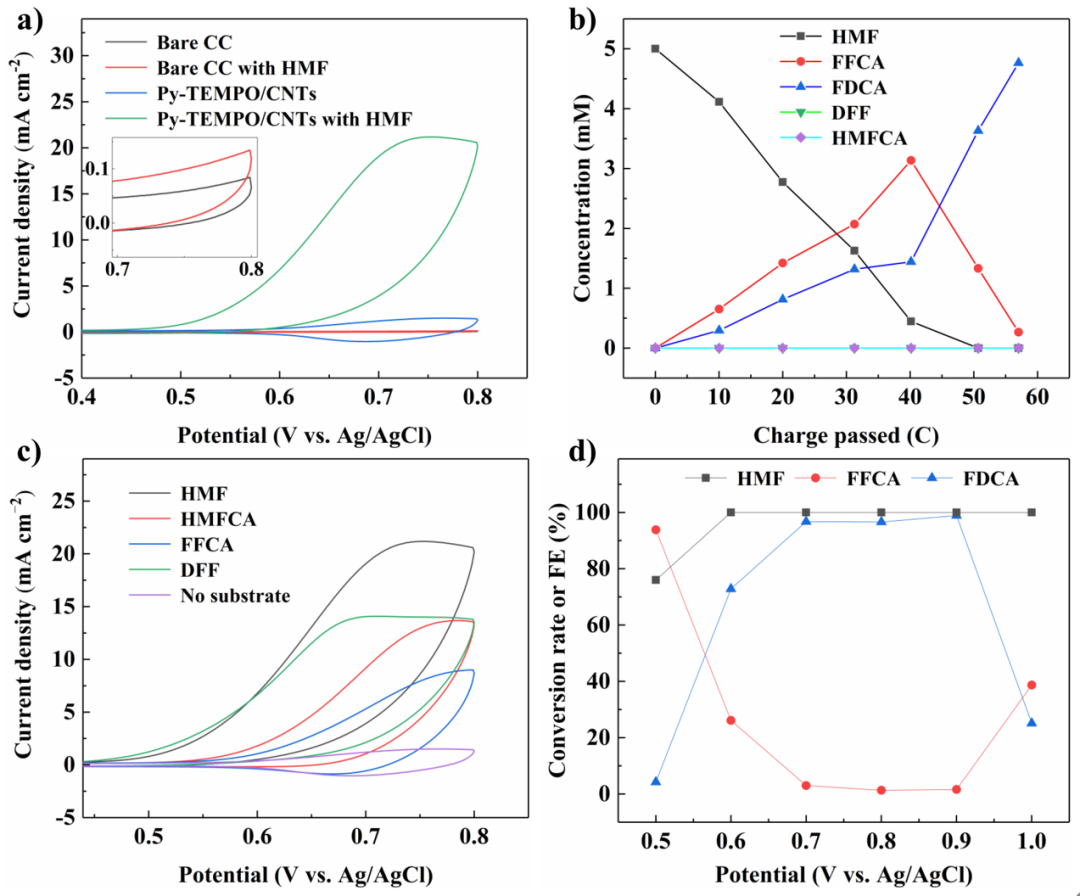
Fig. 3 Electrochemical tests with the Py-TEMPO/CNTs catalyst for the HMFOR. a) CV curves of Py-TEMPO/CNTs with or without 5 mM HMF as well as the blank control groups with only a bare carbon cloth support in 0.2 M Na2CO3 aqueous solution (pH 11.2). Scan rate: 100 mV s−1. b) The concentration of different species during CPE at 0.7 V vs. Ag/AgCl with initial 5 mM HMF in 0.2 M Na2CO3 aqueous solution. c) CV curves of Py-TEMPO/CNTs with 5 mM different substrates in 0.2 M Na2CO3 aqueous solution. Scan rate: 100 mV s−1. d) Conversion rate of HMF and FE of FFCA and FDCA after CPE at varying potentials with 5 mM HMF in 0.2 M Na2CO3 aqueous solution. The CPE tests were performed with a total passing charge of 57.9 C.
在分别研究CO2RR及HMFOR这两个独立的过程之后,我们在三电极体系中进行了两者的耦合电解。为以高FE值获得CO产物,首先控制阴极的CO2RR电位为−1.5 V进行电解实验,发现随着反应进行,阳极电位逐渐增高,引发了其他竞争的氧化进程,导致FDCA的FE值仅为64.8%。而在控制阳极HMFOR电位为0.7 V进行电解时,其初始阴极电位过低,导致阴极反应以析氢为主。而通过增大阴极面积的方式可以有效缓解极化过程,进而使阴极的电位处在恰当的水平以使。以上实验说明了耦合电解过程中不可控的电位变化是影响耦合电解效率的重要因素。

Fig. 4 Paired electrolysis with the potential of the ECO2RR at −1.5 V vs. Ag/AgCl. a) Diagram of the anode potentials and percentage of stoichiometric charge (the ratio of charge passed to the stoichiometric charge 57.9 C) over time. b) Faradaic efficiency of the products in the paired electrolysis.
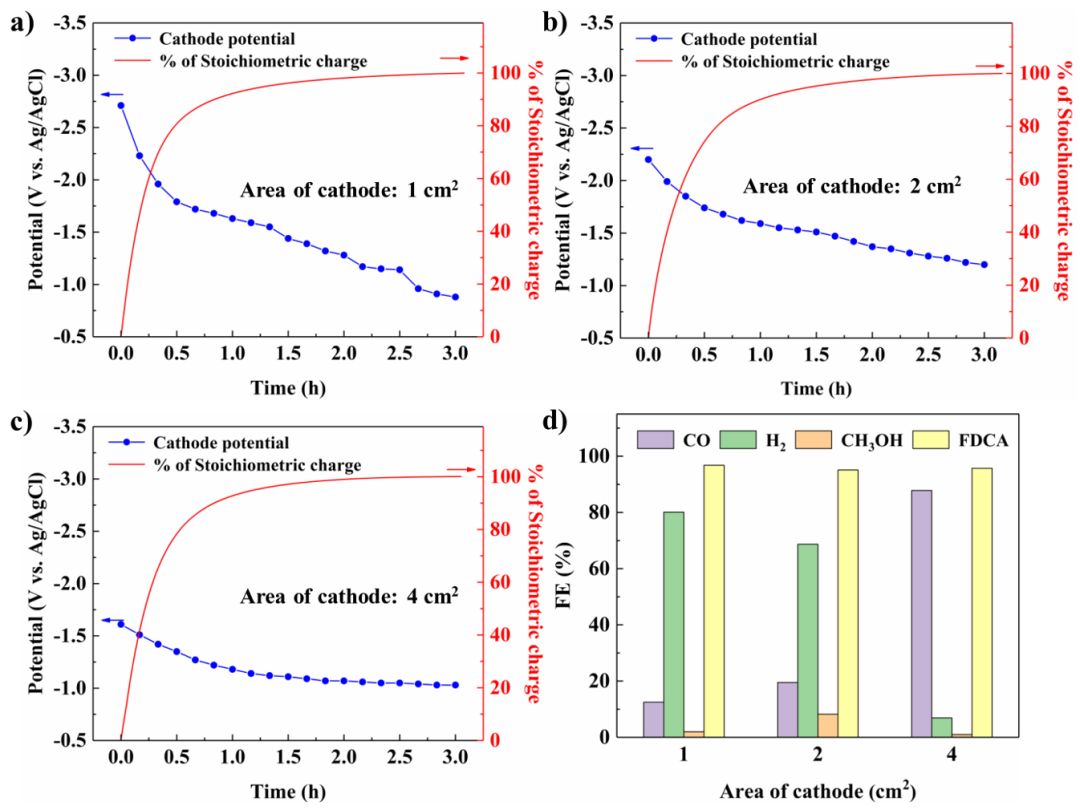
Fig. 5 Paired electrolysis with the potential of the HMFOR controlled at 0.7 V vs. Ag/AgCl. a–c) Diagram of the cathode potentials and percentage of stoichiometric charge over time with cathode areas of 1 cm2, 2 cm2 and 4 cm2. d) The product distribution at varying cathode areas
除三电极系统外,电解槽还可以在双电极配置中完全由电解槽电压控制,这是一种更实用的工业电解模式。然而,在这种情况下,阴极和阳极的电位都不能固定在特定值。随着电解槽压增加,化学计量电荷通过的时间变短,电流密度增大,阴极和阳极的过电位都会有所增加。在2.5 V、3.0 V、3.5 V的槽压下,阴极电位均能保持在−1.3 V到−1.5 V的范围内,使CO的FE值超过95%,当槽压为2.5 V时,阳极电位由在0.45 V ~ 0.87 V的区间内,产生FDCA的FE为90.8%。而在槽压为3.0 V和3.5 V的条件下,FDCA的阳极电位在反应后期均达到0.96 V和1.01 V,使得FDCA的FE值分别下降值86.9%和69.5%。
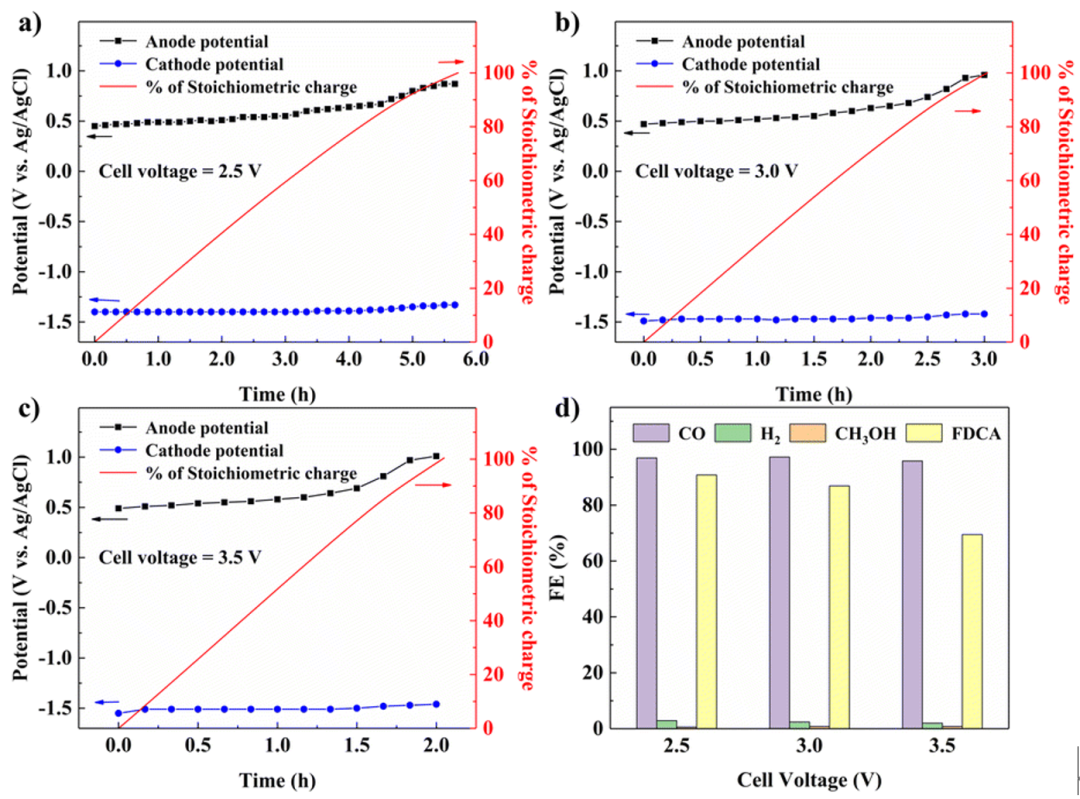
Fig. 6 Paired electrolysis with controlled cell voltages. a–c) Diagram of the potentials at both the anode and cathode, as well as the % of stoichiometric charge over time with cell voltages of 2.5 V, 3.0 V and 3.5 V. d) The product distribution at different cell voltages.
太阳光能是理想的清洁能源之一,在报道的直接光催化氧化HMF生成FDCA的过程中,通常需要碱性环境,而二氧化碳分子在这种条件下倾向于形成碳酸盐。光伏驱动电解可以使氧化和还原反应分别发生在两个电极上,而溶液可以通过膜分离以防止阴极的CO2与阳极的碱性环境接触。基于双电极模式的结果,构建了光伏驱动的耦合电解装置。不同于控制电解时槽压稳定不变,光生槽压随着太阳光强度的波动而在3.0 V和3.5 V之间浮动。在长达3 h的反应过程中,阴极电位保持在-1.4 V至-1.5 V之间,故最终CO的选择性高达95.4%,而在阳极反应中能够以69.9%的产率获得FDCA产品。
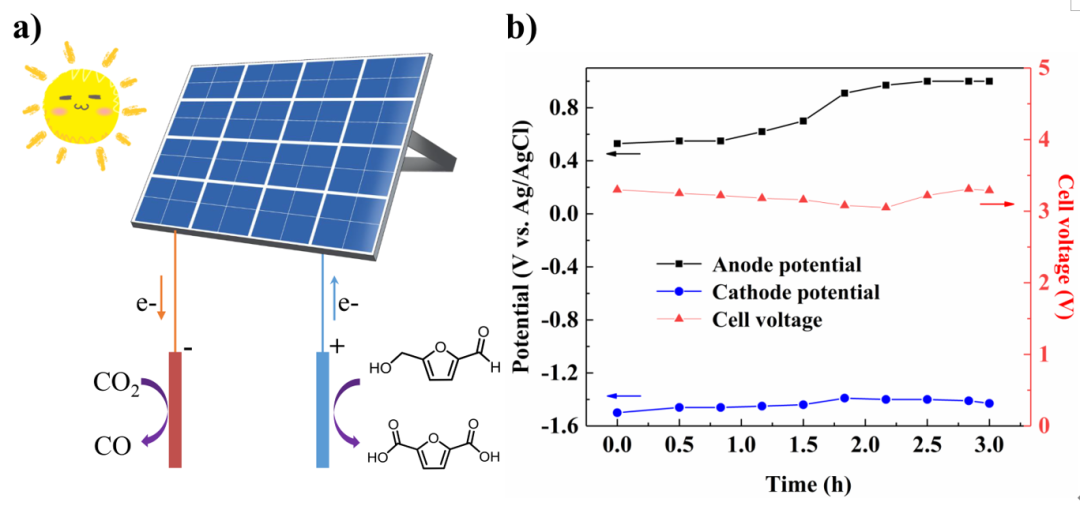
Fig. 7 Sunlight-driven paired electrolysis of the CO2RR and HMFOR. a) Schematic diagram of the system. b) Monitored cell voltage, anode and cathode potentials during this photo-electrolysis procedure.
启发与展望
我们基于两种非共价固定的分子催化剂TBP-CoPc和Py-TEMPO,成功地开发了一种耦合电解体系,在此体系中,HMF氧化产生的质子和电子可用于将CO2还原为CO,同时生成高附加值的FDCA产物。电极面积、电解方法等参数会影响电极的电位,从而影响最终产品的分布。通过对参数的合理调控,可使FDCA和CO的FE值同时达到90%以上。该耦合系统也可采用光伏电源驱动,具有广阔的应用前景。此外,还有一些影响耦合电解反应效率的关键参数,如膜种类及溶液电阻,我们实验室正在进一步研究。
课题组介绍
何良年教授课题组现有研究人员15人,包括教授1名,专任教师3名,博士研究生5名,硕士研究生6名。课题组的主要研究领域包括二氧化碳资源化利用、绿色化学以及生物质转化等领域研究。CO2的捕集与资源化利用,有利于缓解对化石资源的依赖,贡献于可持续发展。因此,探索CO2活化与转化反应,在实现其减排目标的同时还可实现其资源化利用,对于部分替代化石原料的绿色生产方式及实现可持续发展具有重要意义。以促进碳中性为目标,课题组自2003年成立以来,专注二氧化碳化学理论研究与碳捕集利用的新技术开发工作,致力于发展基于催化活化的低能耗、CO2高值化的方法与新途径,如原位催化转化、还原功能化等策略;开发原子、步骤经济性的新反应以及制备重要化学品、平台分子/大宗化学品的绿色合成方法,并为构建杂环化合物(药物分子)提供新思路。通过光及可再生电催化还原, 以克服转化中的能源问题, 开辟利用太阳能光解水制氢与转化CO2相结合的新途径, 即可再生能源储存和CO2转化的可持续发展模式。
课题组网页 http://greenchem.nankai.edu.cn/
何良年教授简介

南开大学教授,博士生导师。1983年毕业于汉江师范学院,1988年9月至1991年6月在华中师范大学攻读硕士学位,师从张景龄教授;1993年9月至1996年6月攻读博士学位期间,在陈茹玉院士指导下从事有机磷杂环化学研究,1996年获理学博士学位。1996年10月-1998年10月,在武汉大学做博士后,从事高分子药物控制释放研究;合作导师:卓仁禧院士。1999年3月-2003年3月,在日本产业技术综合研究所(National Institute of Advanced Industrial Science and Technology,日本筑波)做博士后(NEDO fellow),从事基于二氧化碳资源化利用主的绿色化学研究;合作导师:Toshiyasu Sakakura 教授。1998年入选湖北省高等学校跨世纪学术带头人。2003年回国,被聘为南开大学教授,2004年4月被批准为博士生导师。1998年入选湖北省高等学校跨世纪学术带头人。2009年楚天学者特聘教授,2011年入选英国皇家化学会会士(FRSC)。2016年担任Bentham 科学出版大使(Bentham Science Ambassador)。
课题组专注绿色化学、二氧化碳资源化利用与可再生碳基能源化学研究,在二氧化碳化学及可再生碳基能源化学领域取得了系列创新成果,编著了《二氧化碳化学》与《绿色化学基本原理》,推动了二氧化碳化学领域的发展。迄今发表研究论文300余篇,编写著作3部及英文书籍章节20部 (ACS book series, John Wiley & Sons, Springer, Elsevier,CRC Press Taylor @ Francis),申请(获得)中外专利30项;受邀在大学、研究机构及国际学术会议上作报告80余次,如,IUPAC 绿色化学、Gordon Research Conference、香山科学会议、双清论坛等。曾获天津市自然科学奖、军队科技进步奖。2015年获天津市劳动竞赛示范岗,2018年获南开大学第八届“良师益友”奖。
2013年参与组织并主持第246次美国化学会年会二氧化碳捕集与转化的专题研讨会。参与发起并组织的“中国化学会首届二氧化碳资源化利用大会”于2019年11月8-10日在南开大学成功举行,并获得2019年中国化学会优秀学术交流组织奖。参与组织ACS National meeting、International Conference on Coordination Chemistry、International Conference on Sustainable Chemistry、中国环境化学大会的“二氧化碳捕集与转化”分会。在Green Chemistry、Current Opinion in Green &Sustainable Chemistry、Green Energy Environment、科学通报、Green Chemical Engineering、Asian Journal of Organic Chemistry等学术期刊上发起组织“二氧化碳化学”专刊。自2003年起,开设了《绿色化学》研究专业选修课,并主讲教育部化学专业《化学类导论》视频公开课中的第五讲“绿色化学与可持续发展”。
担任“Green Chemistry and Sustainable Technology” (Springer) Series Editor, Current Organic Synthesis 主编(2015-2021),Frontier in Chemistry 副主编,Journal of CO2 Utilization (Elsevier),ChemistryOpen (Wiley), 科学通报,Current Opinion in Green and Sustainable Chemistry (Elsevier),Mini-review in Organic Chemistry,Green Chemical Engineering等学术期刊编委。