南开大学何良年课题组J. Mater. Chem. A:动态配位提升COF材料光催化CO2还原性能
推文作者:何良年教授团队
第一作者:张永康
通讯作者:何良年教授
通讯单位:南开大学元素有机化学国家重点实验室
论文DOI:10.1039/d4ta07403c

全文速览
共价有机框架(COF)作为一种多孔晶体材料,为光催化CO2还原反应(PCO2RR)提供了一个多功能平台。然而,缺乏表面氧化还原活性位点和快速的光生电荷复合是限制PCO2RR活性进一步提升的主要障碍。在此,本工作设计了一种由动态D··M-A结构COF构建的新型光催化体系。首先,将丰产金属铁位点嵌入到含有联吡啶单元的三嗪基COF结构中(Fe-bpy-COF),随后向体系中补充吡啶硫醇即可实现有效地CO2光还原,整个过程不需要添加额外的光敏剂。值得注意的是,Fe-bpy-COF体系在可见光照射下实现了4052μmol g-1 h-1的甲酸盐收率以及2123μmol g-1 h-1的CO收率。基于1H NMR滴定实验和稳态/瞬态吸收光谱,揭示了优异的光催化性能归因于吡啶硫醇配体与Fe-bpy-COF主体之间的动态配位相互作用。该过程促进了吡啶硫醇配体在可见光下向铁金属中心的连续双电子转移,并抑制了Fe-bpy-COF中的光生电荷复合。最后,结合实验结果和密度泛函理论研究,阐明了还原性铁活性物种催化CO2转化的反应途径。为金属化COF光催化剂的设计以实现更有效地CO2光还原提供了新的研究思路。
背景介绍:
利用太阳能将二氧化碳转化为化学物质和能源小分子可以减轻化学生产对化石能源的依赖,也可以缓解温室效应。目前已经设计和开发了许多用于光催化二氧化碳还原反应(PCO2RR)的有效分子系统。但由于催化剂和光敏剂的分解、二聚化和/或聚集导致失活,分子催化系统通常不太稳定。同时也很难从均相体系中回收溶解的催化剂,这进一步限制了分子催化剂的应用。因此,将具有高活性和选择性的分子活性物种固定到半导体或周期性骨架中被认为是构建实用PCO2RR系统的一种有前景的途径。共价有机框架(COF)作为一类新兴的有机多孔材料,由于其周期性结构、丰富的孔隙率、扩展的π共轭框架、优异的可见光吸收能力和光化学稳定性,为非均相光催化提供了一个有潜力的平台。现已经证明将分子催化剂嵌入三嗪COF骨架中是实现稳定PCO2RR的可行方法。例如,在三嗪-COF合成后通过改性后处理方式将Re(bpy)(CO)3Cl引入到COF中可以有效地实现CO2光还原,具有高选择性和稳定性,CO收率高达750μmol g-1 h-1。尽管如此,快速的光生电荷复合仍然是进一步提高COF光催化效率的主要限制因素。
本文亮点
1. 该系统实现了快速的电子转移,并通过吡啶硫醇配体在Fe-bpy-COF内的往返穿梭有效地抑制了光生电荷复合。配位相互作用促进了电子从吡啶硫醇转移到Fe-bpy-COF中,而动态特征抑制了电子反向转移。
2. 目前,在PCO2RR体系中已经报道了两种主要的单电子转移机制,即还原淬灭和氧化淬灭。本工作提出了一种动态D··M-A结构COF光催化体系中的连续双电子转移机制。该机制主要归因于吡啶硫醇本身的强σ-供体性质及其激发态的强还原性。
3. 该体系在可见光照射下实现了高效的PCO2RR制备甲酸盐和CO。甲酸盐的收率为4052μmol g-1 h-1,CO的收率为2123μmol g-1 h-1。此外,即使在四次循环后,光催化剂仍然表现出优异的PCO2RR性能。
图文解析
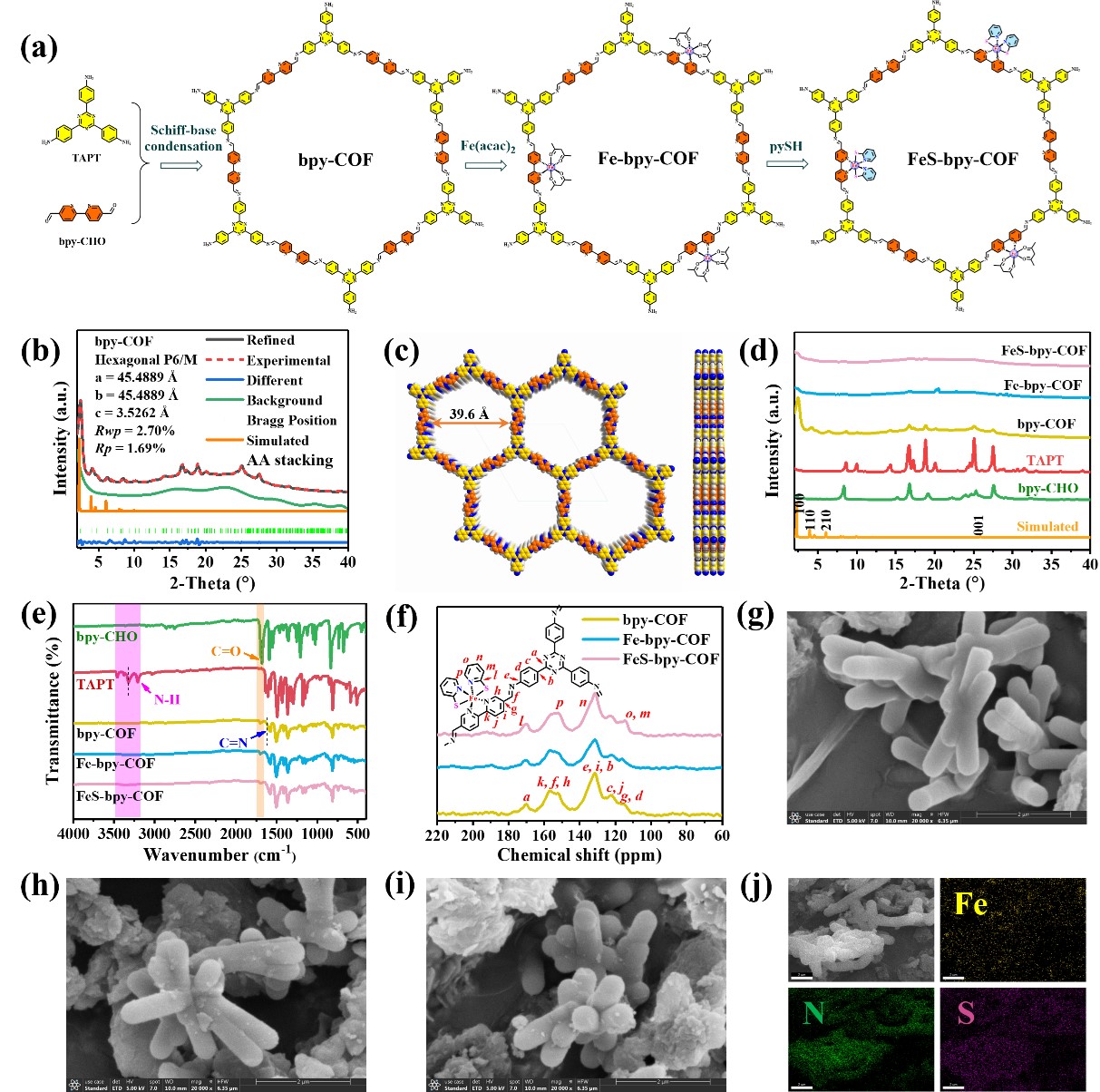
图1 bpy-COF, Fe-bpy-COF和FeS-bpy-COF的制备与结构表征
为了实现将铁吡啶硫醇配合物嵌入COF单元,首先,通过Schiff-base缩合法合成了含有联吡啶单元的三嗪COF(表示为bpy-COF)。随后通过后浸渍法将Fe金属中心整合到bpy-COF-平台中,以提供Fe-bpy-COF。然后,利用配体交换方法将pySH引入Fe-bpy-COF结构中,制备FeS-bpy-COF(图1)。通过一系列结构表征测试证明bpy-COF, Fe-bpy-COF和FeS-bpy-COF的逐步制备。元素mapping图像显示在FeS-bpy-COF中Fe, N, S元素是均匀分布的(图1j)。
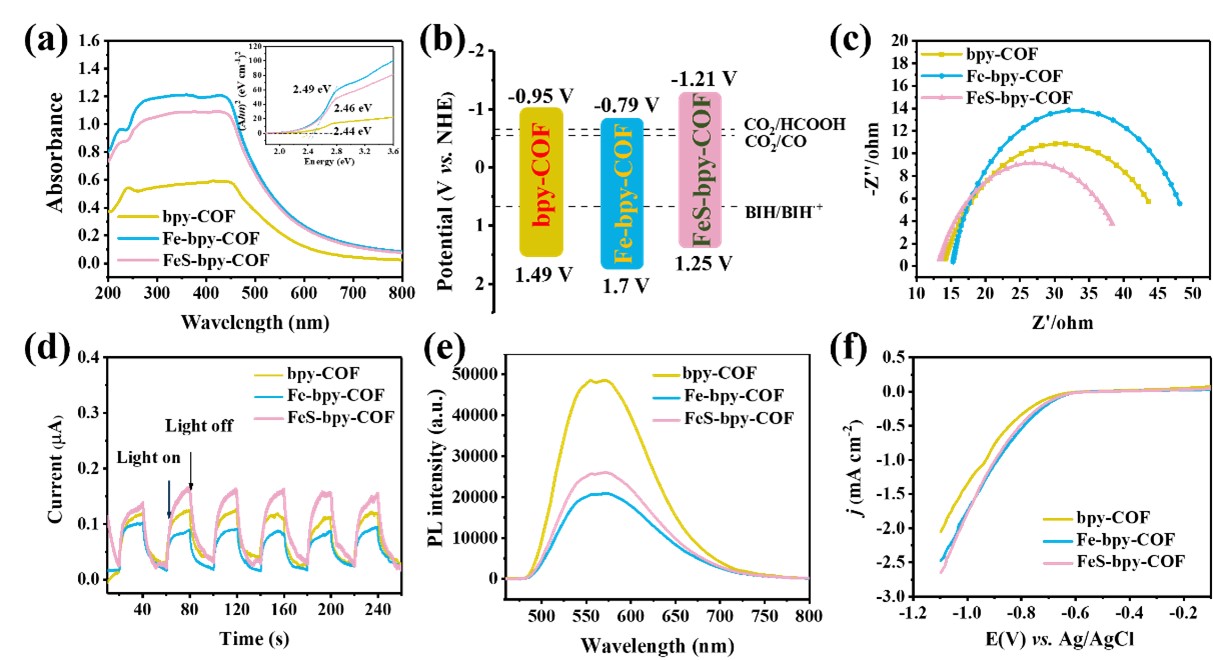
图2 bpy-COF, Fe-bpy-COF和FeS-bpy-COF的光电性能表征
为了研究这三种材料的光电性能,进行了一系列光电化学测试。紫外-可见吸收光谱显示,Fe-bpy-COF和FeS-bpy-COF的光吸收能力高于bpy-COF(图2a)。根据Tauc图发现这三种结构具有相似的带隙。与bpy-COF(2.44eV)相比,Fe-bpy-COF(2.49eV)和FeS-bpy-COF(2.46eV)的带隙能量略宽(图2a,插图)。随后结合Mott-Schottky方程计算获得了bpy-COF、Fe-bpy-COF和FeS-bpy-COF三种材料的能带结构(如图2b所示),证实了所有材料理论上均符合将二氧化碳还原为HCOOH和CO的热力学要求。特别是,FeS-bpy-COF具有更高的导带电位,预计更有利于二氧化碳的还原。同时更小的电化学阻抗,更高的光电流响应以及较低的荧光发射强度提供更有利的光催化条件(图2c-e)。在CO2气氛下进行的线性扫描伏安(LSV)测试结果表明(图2f),与 bpy-COF 相比,Fe-bpy-COF和FeS-bpy-COF都表现出更高的催化电流密度,说明铁金属中心可能作为促进 CO2还原的活性位点。
利用飞秒瞬态吸收光谱(fs-TAS)研究了光生载流子的动力学。如图3所示,经370nm激光闪光激发后,在400~700nm处有明显的正带吸收,属于激发态吸收(ESA)。由于重叠的ESA信号覆盖,没有观察到显著的基态漂白和受激发射信号峰。所有样品的弛豫过程都发生在皮秒范围内(图3d),FeS-bpy-COF样品表现出最长的弛豫时间(τavg=415ps),这表明铁离子和pySH配体的掺入抑制了FeS-bpy-COF中的光生电荷复合。
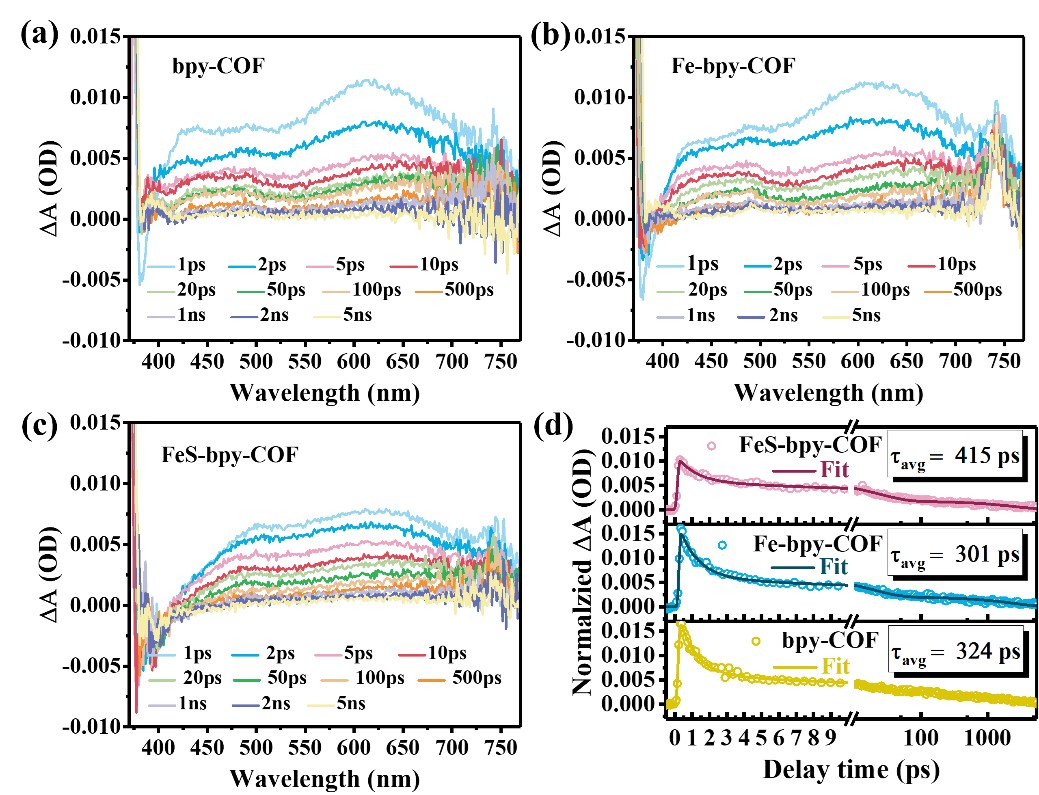
图3 bpy-COF, Fe-bpy-COF和FeS-bpy-COF的飞秒瞬态吸收光谱
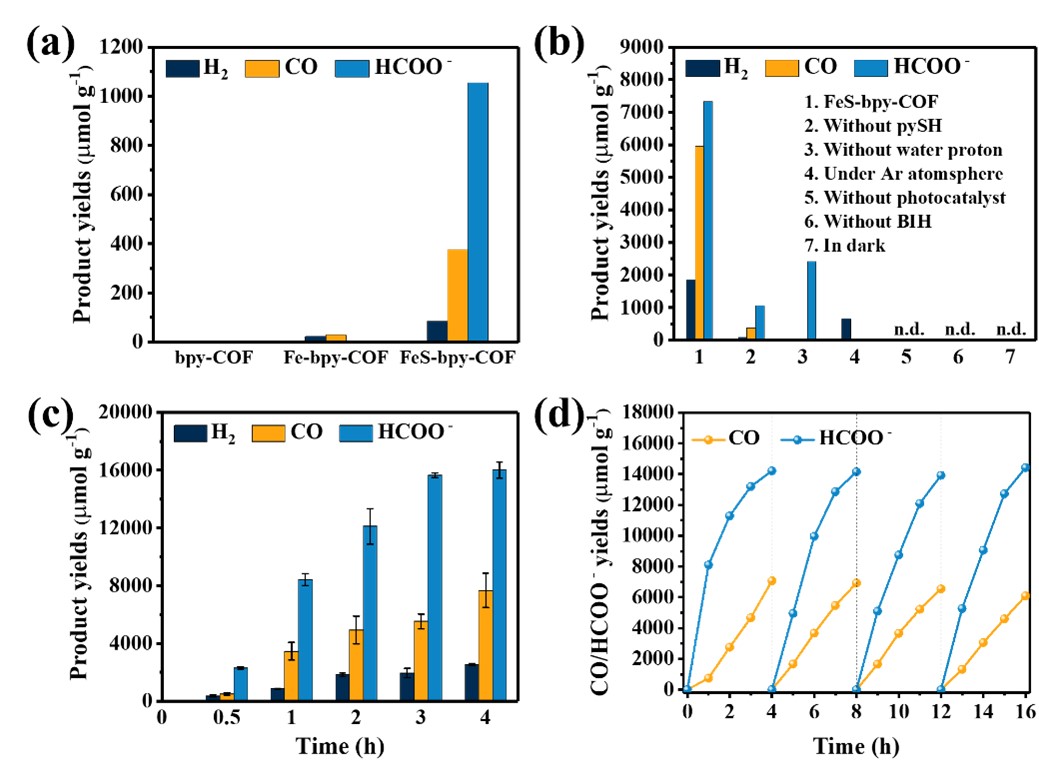
图4 bpy-COF, Fe-bpy-COF和FeS-bpy-COF的光催化CO2还原性能测试
在300W Xe灯(λ≥400nm)照射下评估了三种材料的光催化CO2还原性能。在该系统中,H2O用作质子源,BIH用作电子牺牲剂,不添加任何额外的光敏剂。如图4a所示,在连续照射4小时后,bpy-COF没有表现出任何PCO2RR活性,而Fe-bpy-COF产生少量CO,产量为30 μmol g-1,并伴有24μmol g-l的H2释放。使用FeS-bpy-COF代替Fe-bpy-COF作为光催化剂,PCO2RR性能提高了约48倍。值得注意的是,添加额外的pySH进一步提高了活性。通过对照实验以确定CO2光还原的关键因素(图4b),在没有光、BIH、光催化剂或Ar气氛的情况下,没有检测到CO2还原产物。在没有H2O作为质子源时,催化性能显著降低,只检测到少量甲酸盐产物。在0.4 mM pySH的存在下,Fe-bpy-COF表现出优异的PCO2RR活性(图4c)。光照4小时后甲酸盐产量达到16207μmol g-1,CO产量达到8491μmol g-l。另外,该催化剂在四个循环后仍保持优异的CO2光还原性能(图4d)。
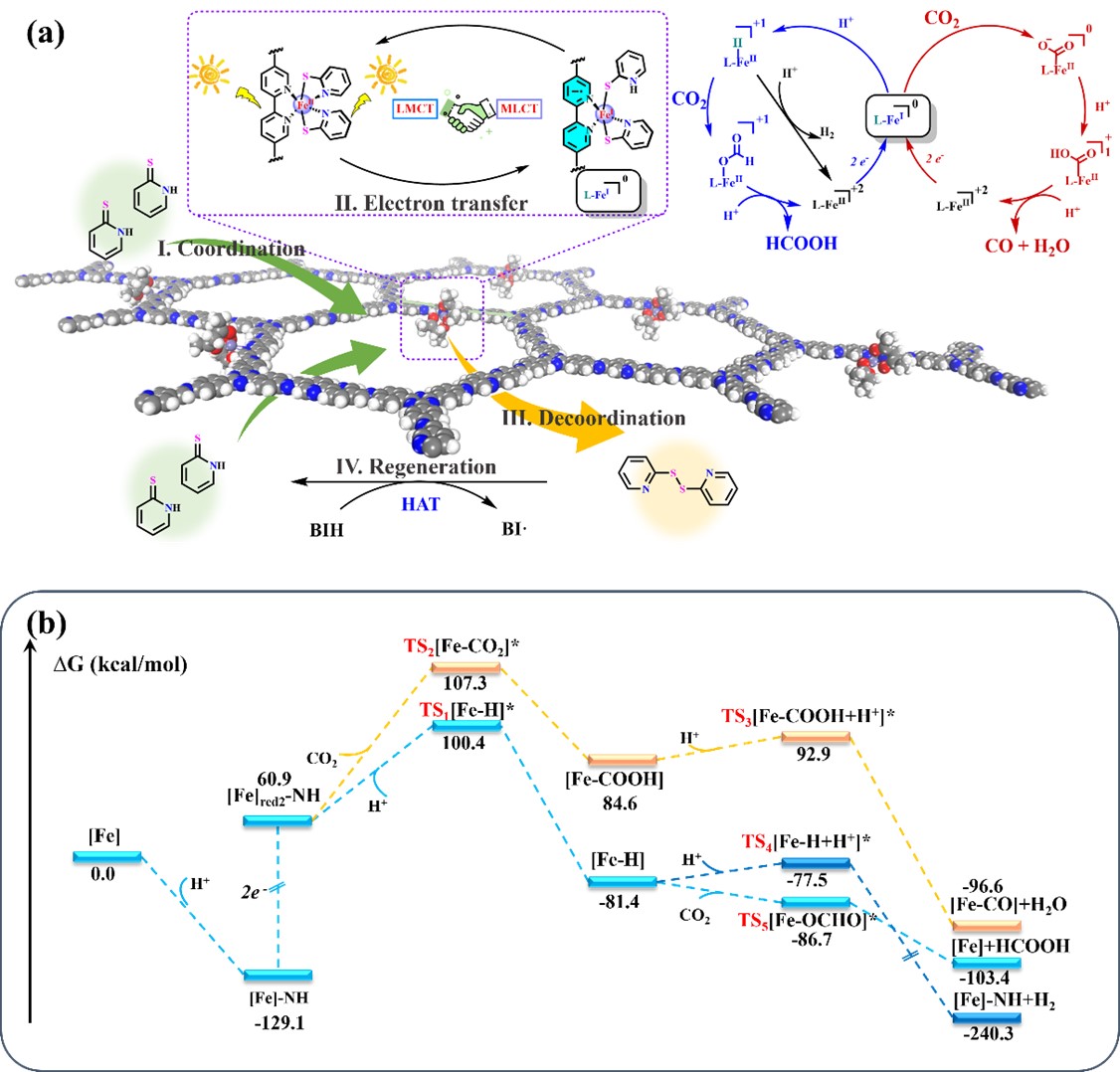
图5 可能的电子转移途径以及光催化CO2还原理论计算结果
如图5a所示,本工作提出了动态D··M-A结构中可能的电子转移途径。这项工作中的动态特征主要涉及四个过程:配位、电子转移、脱配位和再生。游离的pySH配体与Fe中心配位后吸收可见光,同时通过LMCT将两个电子转移到Fe(II)中心,形成潜在的Fe(0)物种。该物种在可见光照射下通过MLCT快速将电子转移到COF骨架。在转移电子后两个硫醇配体脱配位并形成二硫二吡啶,随后通过与BIH的氢原子转移反应再生pySH。独特的电子转移模式有效地绕过了连续还原引起的高能势垒(图5b),形成的[Fe]red2-NH物种可以直接活化CO2形成[Fe-COOH]中间体,也可以结合质子形成[Fe-H]物种。直接活化CO2的反应能垒(∆G‡ = 46.4 kcal/mol)略高于形成[Fe-H]物种的反应能垒(∆G‡ = 39.5 kcal/mol),表明CO的产生不是主要途径,这与实验结果一致。在主要的反应途径中,[Fe-H]物种更倾向于进行CO2插入反应以形成甲酸盐而不是进一步结合质子产生氢气,这也与实验结果中获得的较高甲酸盐选择性相一致。
总结与展望
综上,本工作开发了一种新型的COF光催化体系,该体系由动态D··M-A结构构建而成,用于高性能的CO2光催化还原。具体来说,该系统通过在Fe-bpy-COF中pySH配体的往返穿梭实现快速电子转移并有效地抑制光生电荷复合。结果表明,该体系在可见光照射下实现了有效的PCO2RR生成甲酸和CO。另外,四次循环后,Fe-bpy-COF光催化剂仍然表现出优异的PCO2RR性能。为COF光催化剂的结构设计提供前所未有的研究思路,并为目前光催化系统中的电子转移途径提供新的见解。
文献信息
Dynamic Pyridinethiol Ligand Shuttling within Iron-Anchored Covalent Organic Frameworks Boosts CO2 Photoreduction
Yong-Kang Zhang,aLan Zhao,aAlexander O. Terent’ev,b Liang-Nian He*a
J. Mater. Chem. A, 2024,DOI: 10.1039/d4ta07403c
https://doi.org/10.1039/D4TA07403C
图文摘要/TOC图
课题组介绍
何良年课题组介绍
何良年课题组在CO2转化策略的设计和基于活化机理的高效催化剂开发方面做了系统的工作,不仅提出了碳捕集与转化耦合、CO2分级可控还原功能化、利用多组分串联反应突破热力学限制及光促进的CO2转化等策略,还针对不同的策略开发出了相应的高效催化体系,实现了温和条件下CO2的转化,取得了多项创新性成果。何良年教授课题组现有研究人员19人,包括教授1名,专任教师3名,博士研究生8名,硕士研究生6名,科研助理1名。目前课题组的主要研究领域包括二氧化碳化学(二氧化碳化学转化方法学)、可再生碳基能源化学、生物质高值化利用、二氧化碳捕集、二氧化碳光电催化/合成。
详见课题组主页http://greenchem.nankai.edu.cn.