英文原题:Nickel-Catalyzed Stereoselective Synthesis of Dihydrobenzofuran Exocyclic Alkenyl Carboxylic Acids from CO2
中文题目:丰产金属催化构建CO2基复杂分子:镍催化以CO2为羧基源的立体选择性合成二氢苯并呋喃丙烯酸衍生物
通讯作者:何良年 (Liang-Nian He)
作者:丁杰,谢汶均,武安国,许立锋,张明,李红茹,何良年 (Jie Ding, Wen-Jun Xie, An-Guo Wu, Li-Feng Xu, Ming Zhang, Hong-Ru Li, Liang-Nian He)
背景介绍
工业化进程在推动社会发展的同时,也导致CO2排放量急剧增加,引发了一系列环境与气候问题。在此背景下,将CO2视为一种廉价、丰富、无毒且可再生的C1合成子,通过催化转化制备高附加值化学品,已成为实现碳捕集与利用(CCU)战略的关键路径之一。在众多CO2高值化转化方向中,羧酸的合成尤为重要,然而现有方法大多局限于结构简单的羧酸产物,限制了其应用范围。另一方面,二氢苯并呋喃(DHB)骨架与α,β-不饱和羧酸结构单元广泛存在于多种天然产物与药物分子中,表现出显著的生物活性。在现代药物研发中,将具有不同生物活性的分子结构进行合理整合,是提升化合物成药性与功能多样性的重要策略。因此,发展高效方法以构建二氢苯并呋喃丙烯酸衍生物,具有重要研究意义与应用前景。在这一方向上,分子内Heck反应因其能够通过“一锅法”实现环状骨架的构建与位点选择性官能团化,为高效合成结构复杂、功能明确的杂环羧酸提供了极具潜力的合成工具。
文章亮点
1. 首次实现了丰产金属镍催化的Heck型环化/羧化反应,使用CO2作为羧基源,合成了具有二氢苯并呋喃丙烯酸衍生物,深入的机理研究阐明了涉及Ni(0)/Ni(II)/Ni(I)的催化循环,并通过对配体结构的系统优化,揭示了促进CO2插入的关键空间与电子效应。
2.高立体选择性:产物具有专一的顺式选择性,实现了立体可控合成。
3. 该策略具有良好的官能团兼容性,克级规模的成功合成展现了应用潜力。此外,二氢苯并呋喃环外烯基羧酸产物可进一步芳构化或与天然产物(如丁香酚)进行酯化,展示了其作为合成砌块的价值。

图1 镍催化以CO2为C1源合成二氢苯并呋喃环外烯基羧酸
图文解读
首先,本研究以碘苯串联炔丙基醚1a为模板底物,对反应条件进行了系统筛选。在配体结构优化过程(图2)中发现,无论使用菲罗啉型还是联吡啶型配体,当氮原子邻位存在取代基时,均未检测到目标产物2a,而是生成质子化副产物2a-H。该结果表明,氮邻位取代基的空间位阻效应会显著抑制CO2对C–Ni键的插入。进一步研究表明,采用氮原子对位为甲氧基取代的联吡啶配体L11可有效提升反应效率,将2a的产率提高至72%,可能是由于L11的给电子效应增强了镍中心的电子密度,进而促进了其对CO2的亲核进攻。除配体外,添加剂与碱的筛选也对反应效率具有关键影响。控制实验表明,MgCl2在本转化中不可或缺,其作用机理包括促进镍催化剂的还原及稳定Ni(I)-CO2加合物,从而加速CO2插入步骤。此外,碱的引入有助于提升溶液中CO2的溶解度,抑制质子化副产物2a-H的生成。在多种碱中,Et3N表现最优。基于上述筛选结果,通过对催化剂前体、还原剂及溶剂等参数的进一步优化,最终确定了该反应的最佳条件:以NiI2为催化剂前体,L11为配体,Mn粉为还原剂,MgCl2为添加剂,Et3N为碱,DMF为溶剂,在CO2气氛下于室温反应10小时。
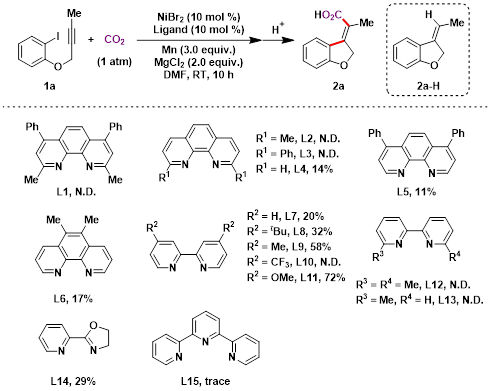
图2 配体效应对碘苯串联炔丙基醚1a参与的Heck型环化/羧化反应的影响
在确定最佳反应条件后,本研究对一系列含不同取代基的碘苯炔丙基醚类底物进行了适用性考察(图3)。结果表明,该反应具有良好的官能团耐受性,可兼容卤素(3f–3h)、酮羰基(2i)、氰基(3j)等对格氏试剂敏感的官能团,为产物的后续衍生化提供了可能。进一步探究芳环上取代基电子效应对反应的影响发现,C6位取代基的性质对反应具有显著影响,具有较弱给电子效应的甲基(3l)相较于强吸电子的三氟甲基(3m)展现出更高的反应活性,而当C6位为强给电子甲氧基时,通过p–π共轭作用稳定芳构化过渡态,最终生成苯并呋喃产物4k。此反应还可以扩展至苯并吡喃类似物3r和3s。为阐明反应的立体化学特征,本研究通过2a的单晶X射线衍射以及3r和3s的NOESY谱图进行分析,明确证实碳碳三键对C-Ni键的迁移插入过程具有顺式选择性,从而从结构上验证了该Heck型环化/羧化反应的高立体控制能力。

图3 碘苯串联炔丙基醚与CO2的Heck型环化/羧化反应的底物范围
为验证本方法的合成实用性,开展了一系列应用研究(图4)。首先,在克级规模实验(4 mmol)中,底物1a顺利转化为目标产物2a,产率为63%。进一步对产物进行衍生化实验显示,2a在Fe(OTf)3催化下可高效发生芳构化反应,以91%的产率得到苯并呋喃衍生物4a,该过程无需额外添加氧化剂或化学计量的碱,步骤简洁、条件温和。此外,2a的羧基可与天然产物丁香酚发生酯化反应,生成未芳构化酯5a与芳构化酯6a的混合物(5a:6a = 1:1.41)。上述结果共同表明,二氢苯并呋喃环外烯基羧酸2a在后期官能化及类药分子构建中展现出良好的应用前景。

图4克级规模制备及衍生化
为深入探究反应机理,开展了一系列机理验证实验(图5)。首先,在使用零价镍络合物Ni(COD)2替代NiI2作为催化剂时,仍能以55%的产率获得目标产物2a,表明Ni(II)催化剂可被Mn还原为Ni(0)物种,进而启动催化循环。在采用化学计量的Ni(COD)2且不添加Mn的条件下,反应未检测到2a的生成;而当向该体系加入3当量Mn后,2a的产率为35%,这一结果说明反应中涉及Ni(I)关键中间体的形成并参与了催化循环。此外,通过13C同位素标记实验验证了羧基的来源,质谱分析显示2a-13C中羧基碳的标记率超过99%,证明产物中的羧基完全来源于反应气氛中的CO2。
基于上述机理实验及相关文献报道,提出了一个可能的催化循环机理(图5)。该反应起始于Ni(II)催化剂前体被Mn还原为活性Ni(0)物种,继而与底物1a发生氧化加成,形成Ni(II)中间体A。随后,反应可能经由两条路径生成关键Ni(I)中间体D:路径I中,Ni(II)中间体A先经历分子内碳碳三键的迁移插入,生成Ni(II)环化中间体B,随后被Mn还原为Ni(I)中间体D;路径II则为Ni(II)中间体A先被Mn还原为Ni(I)物种C,随后发生5-exo-dig环化,同样得到中间体D。接下来,中间体D对CO2进行亲核进攻,实现CO2插入至C–Ni(I)键,形成羧酸镍中间体E。该中间体经Mn还原及转金属化后,生成中间体F,并释放Ni(0)完成催化循环。最后,中间体F经酸化处理或甲酯化,分别得到羧酸产物2a或甲酯化产物3a。

图5 机理探究及可能的反应机理
总结与展望
综上,本文成功开发了一种镍催化的Heck型环化/羧化反应,利用CO2作为C1合成子,一步构建了兼具二氢苯并呋喃骨架与不饱和羧酸结构的复杂分子。该方法具有步骤简洁、立体选择性好等优点,不仅为CO2的资源化利用提供了新策略,也为药物先导化合物中含氧杂环骨架的构建提供了绿色、高效的合成工具。未来,通过对催化体系的进一步优化与拓展,有望将CO2更广泛地应用于具有生物活性复杂分子的精准合成中。
详见课题组主页http://greenchem.nankai.edu.cn
文章链接:
https://doi.org/10.1021/acs.orglett.5c03063